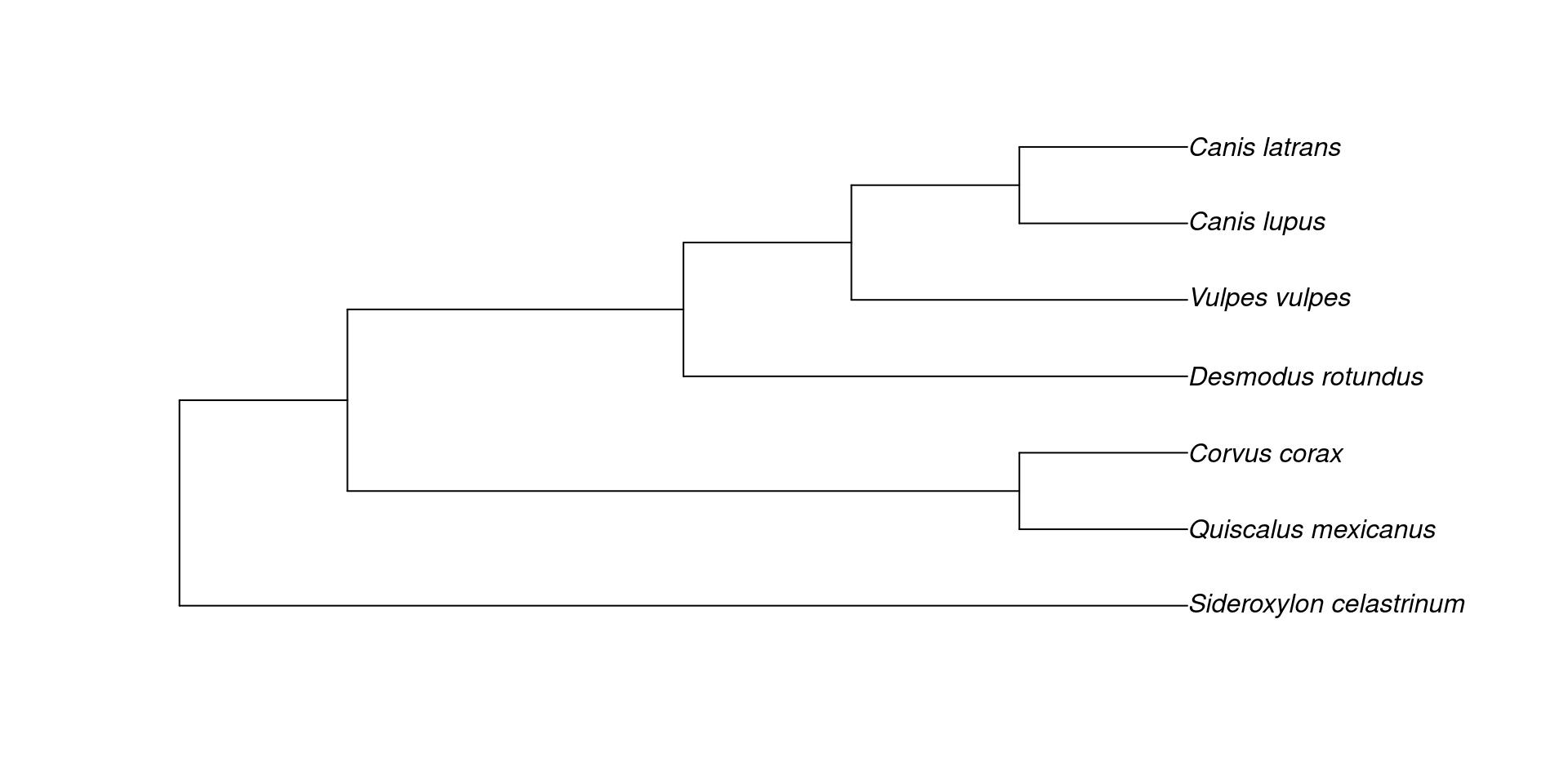
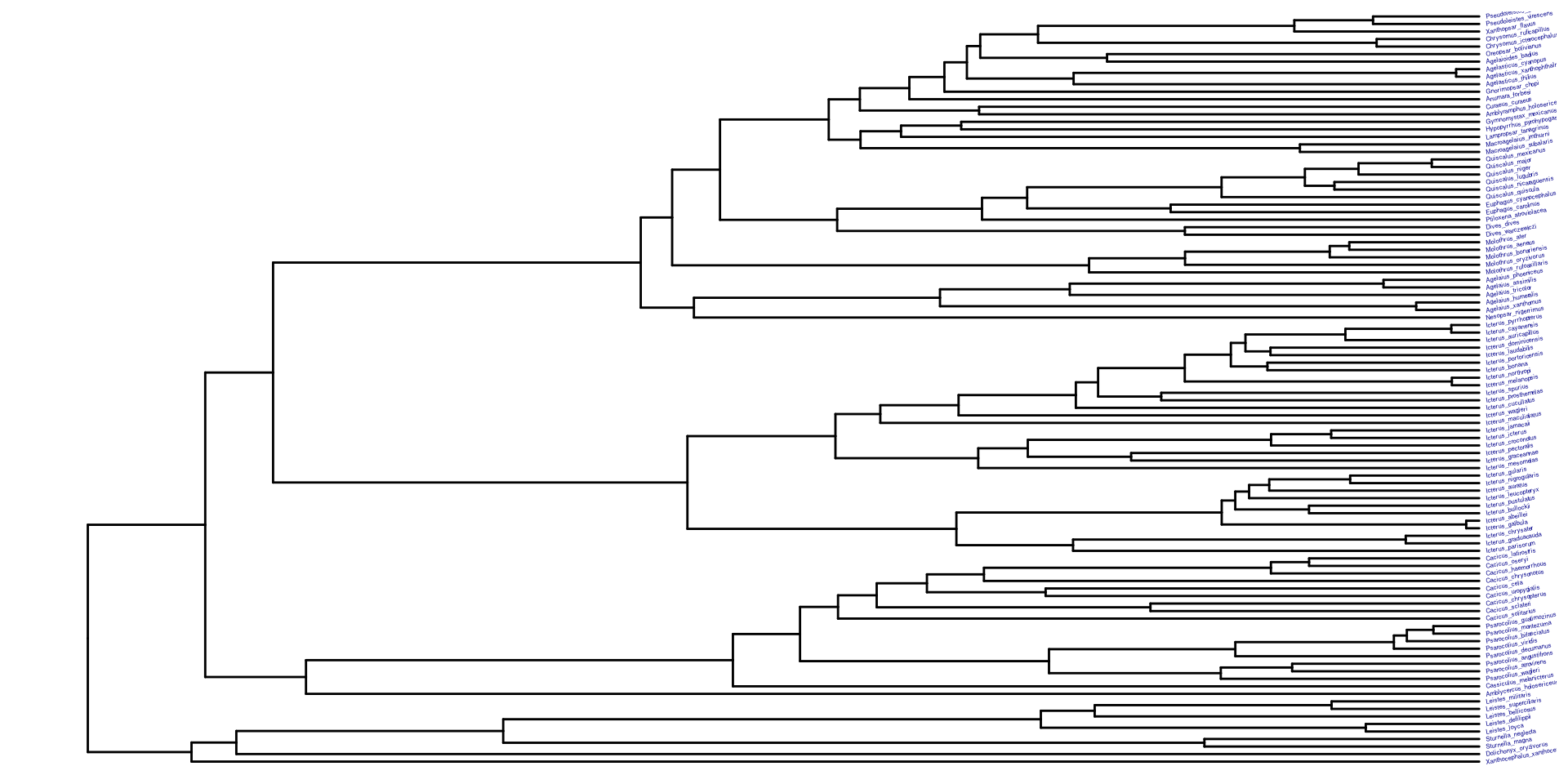
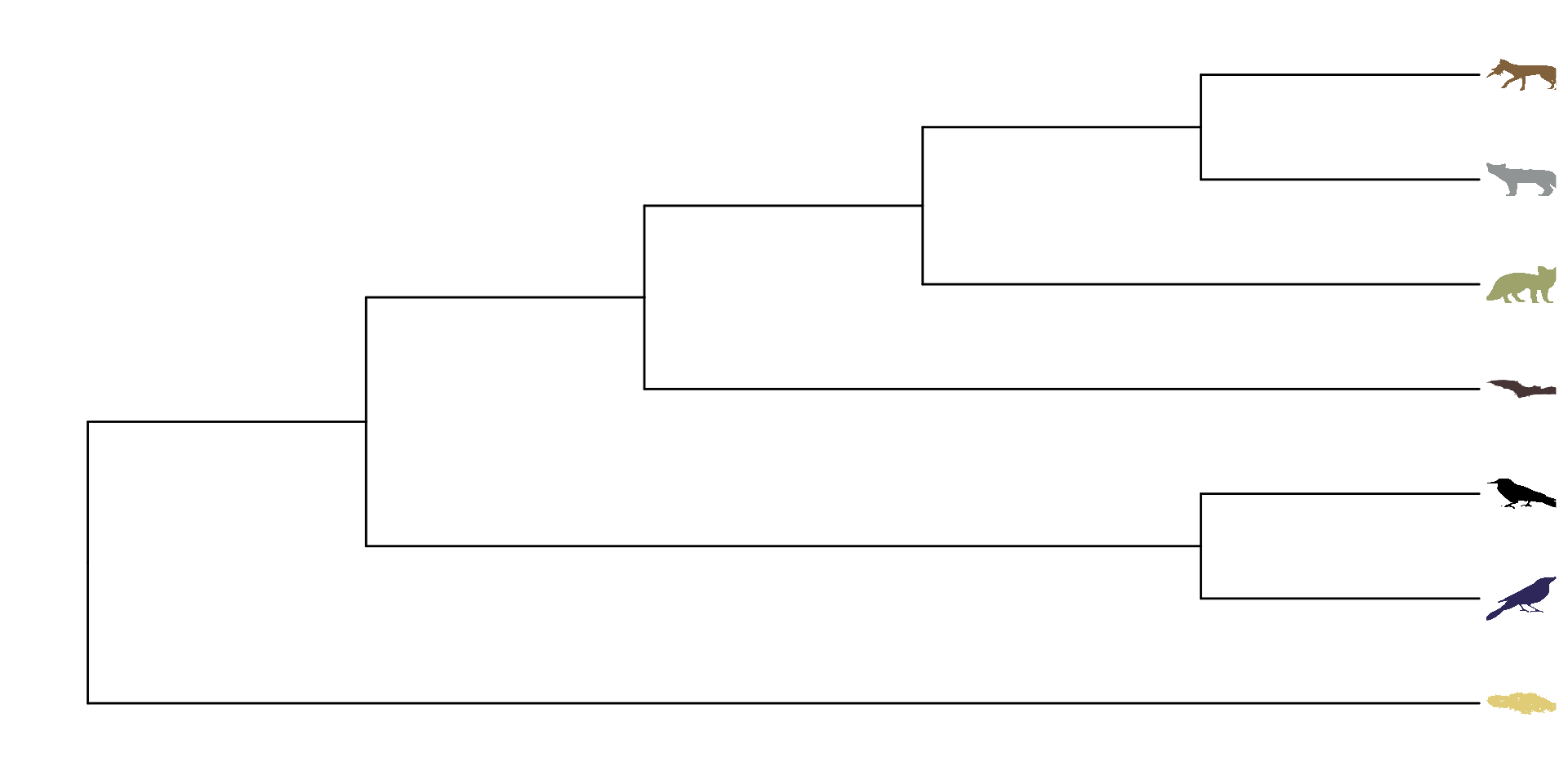
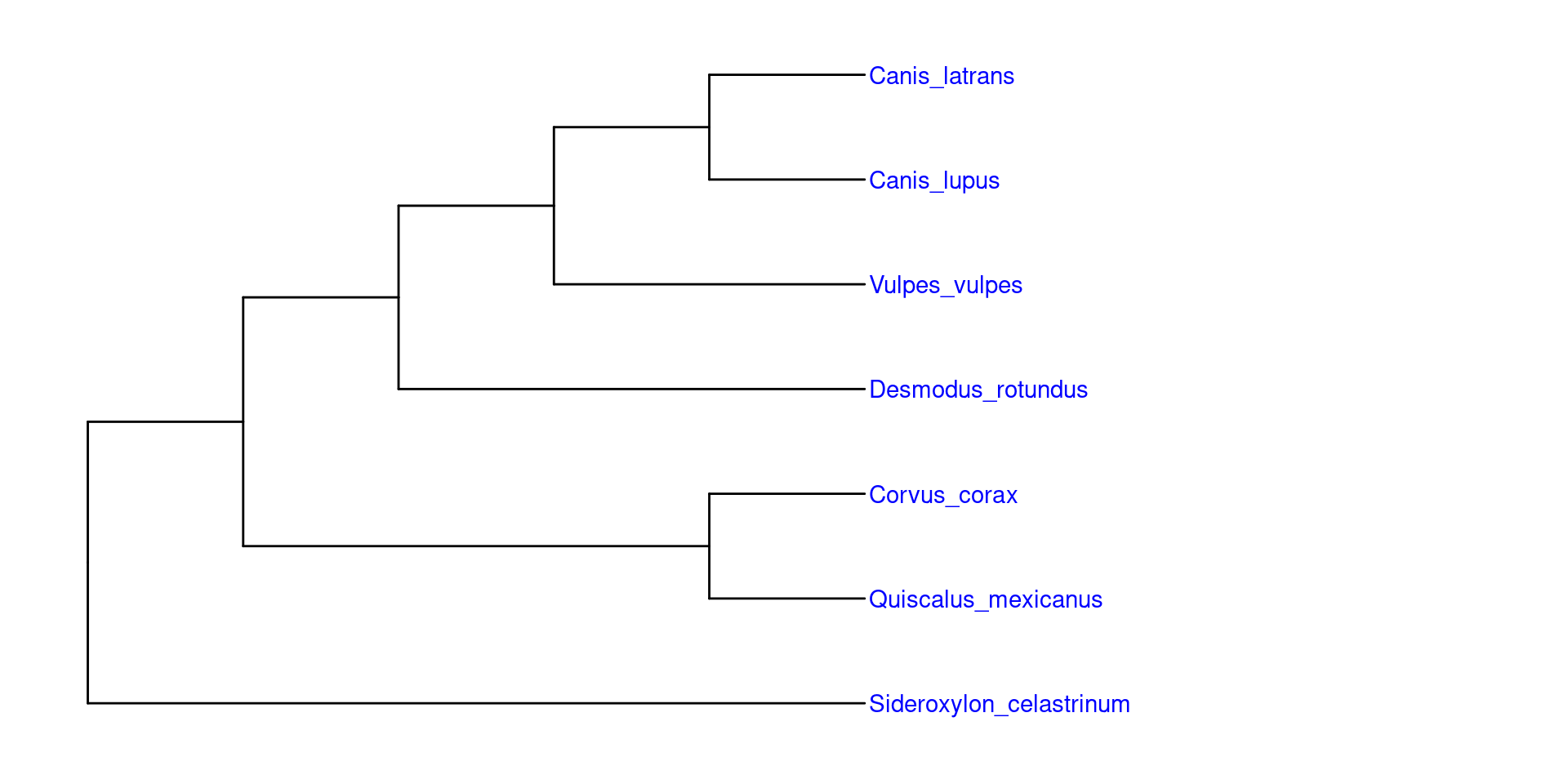
En este caso usaremos un árbol filogenético en particular (la filogenia de Icteridae). Sin embargo, estos ejemplos se pueden hacer en su mayoría con cualquier otra filogenía que tengan disponible.
Phylogenetic tree with 100 tips and 99 internal nodes.
Tip labels:
Icterus_melanopsis, Icterus_northropi, Icterus_laudabilis, Icterus_dominicensis, Icterus_cayanensis, Icterus_pyrrhopterus, ...
Node labels:
761, 762, 763, 764, 765, 766, ...
Rooted; includes branch lengths.
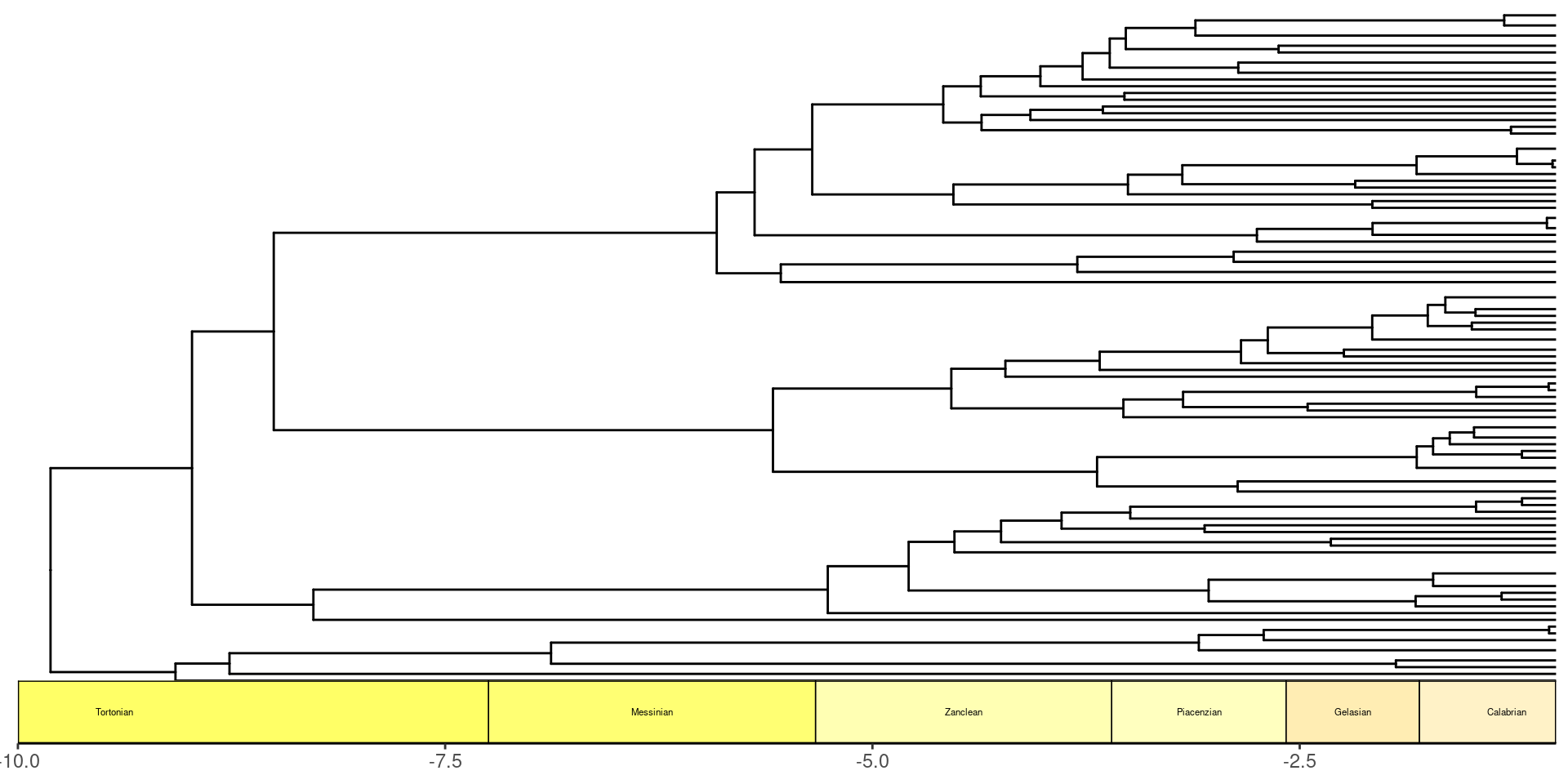
Veamos esta filogenia graficandola usando ape, la manera base para R.
plot(ictree, show.tip.label=F)
Ahora graficaremos el mismo árbol utilizando ggtree, el cual sigue una formula idéntica a la de ggplot. Nuestro árbol base se llamará p1:
p1<-ggtree(ictree,color="black",size=0.5)plot(p1)
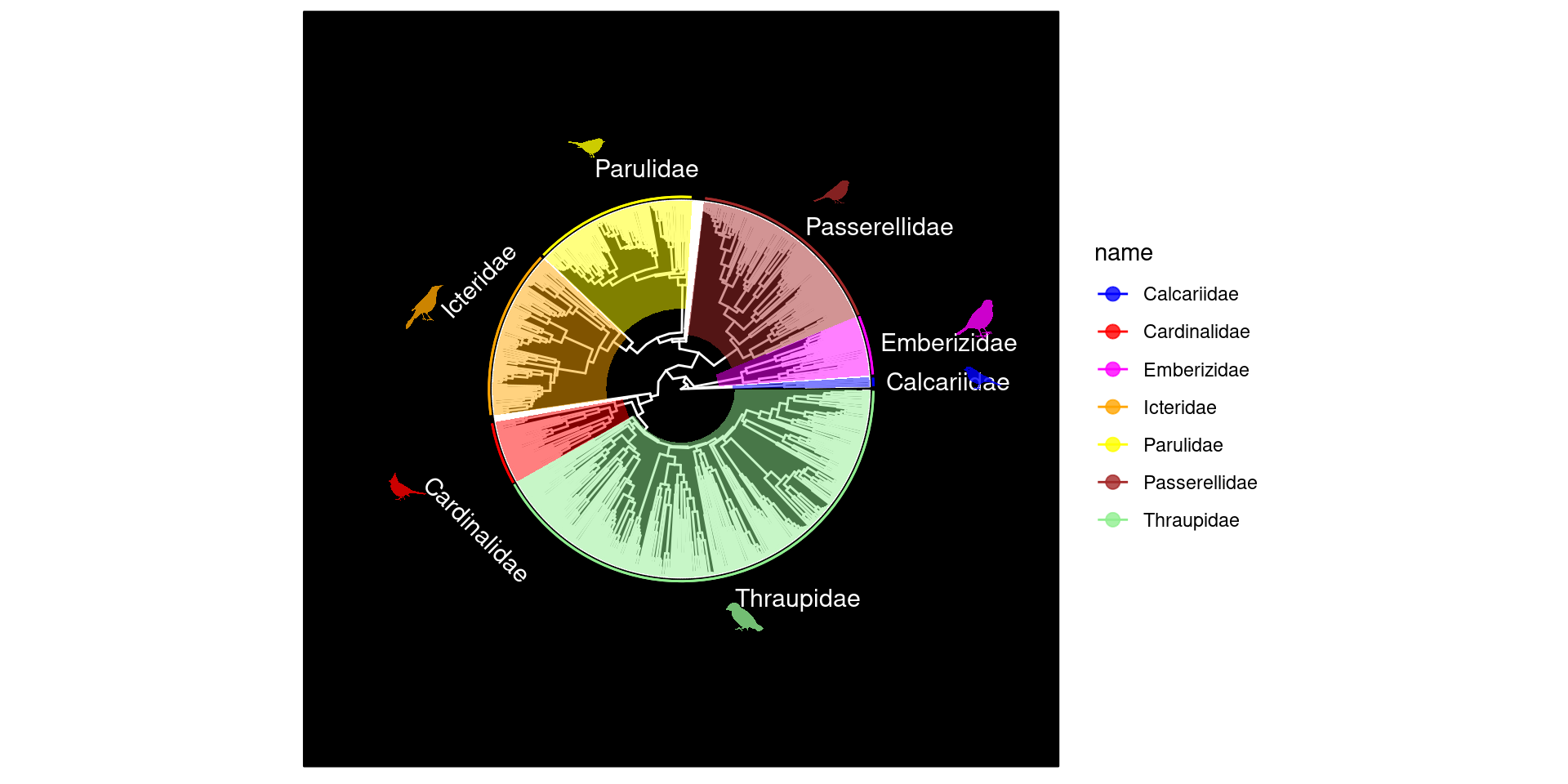
Con p1, al igual que con cualquier otro gráfico de ggplot, podemos agregar parametros de graficación para personalizar o agregar anotaciones a nuestra filogenia. Primero empezaremos con los parametros de personalización. Por ejemplo, podemos cambiar la disposición de la filogenia utilizando el parametro layout_:
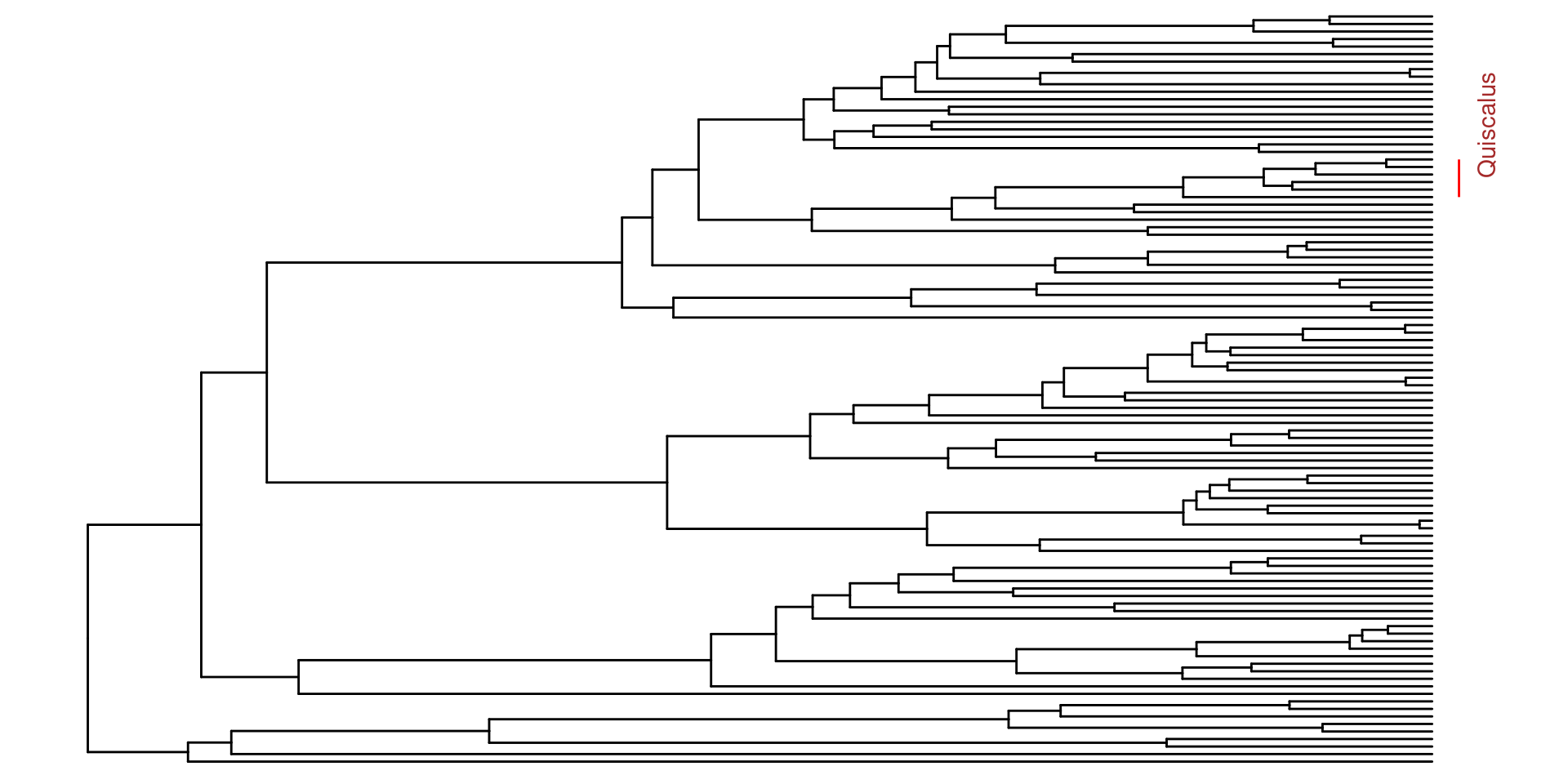
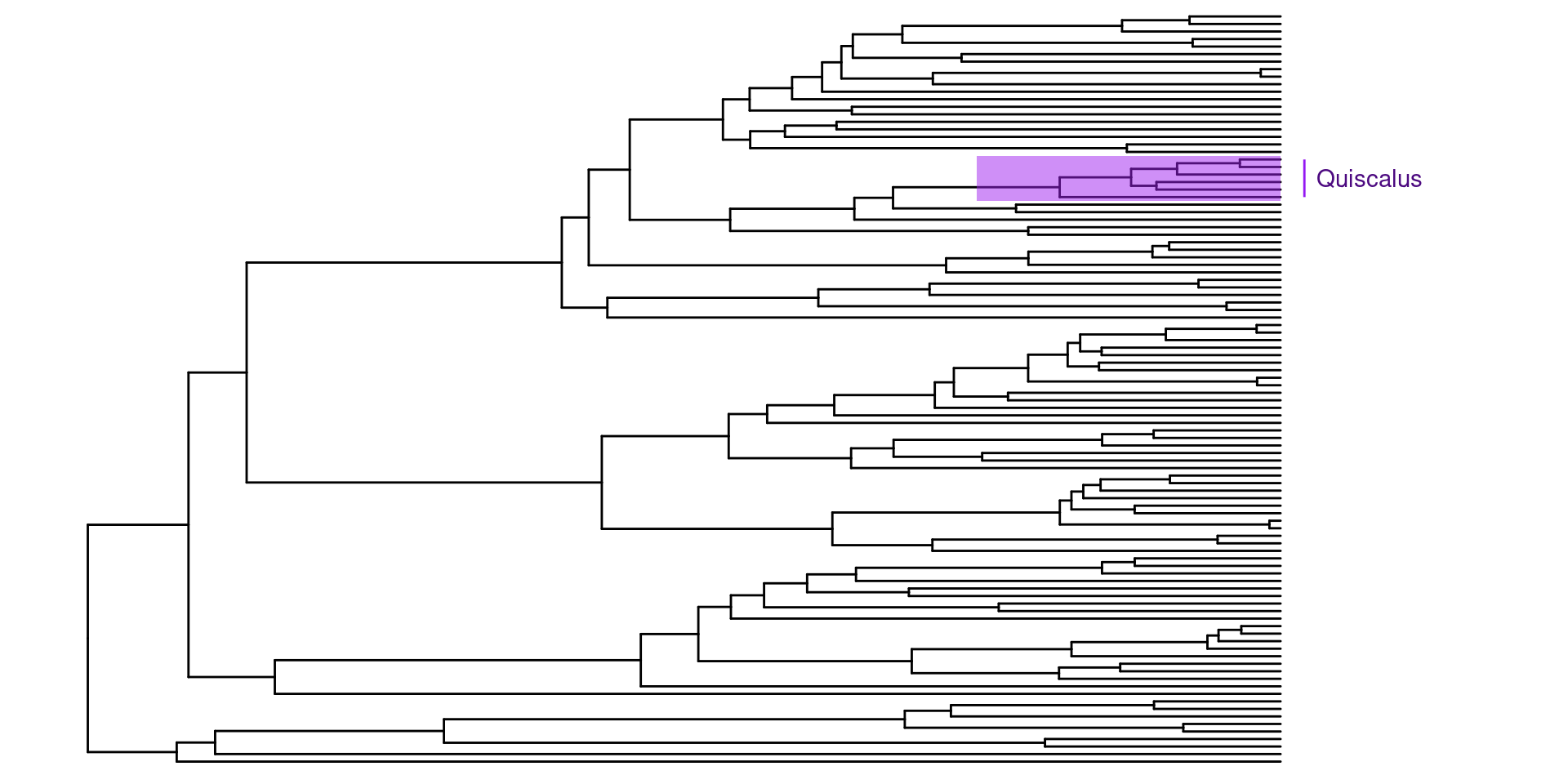
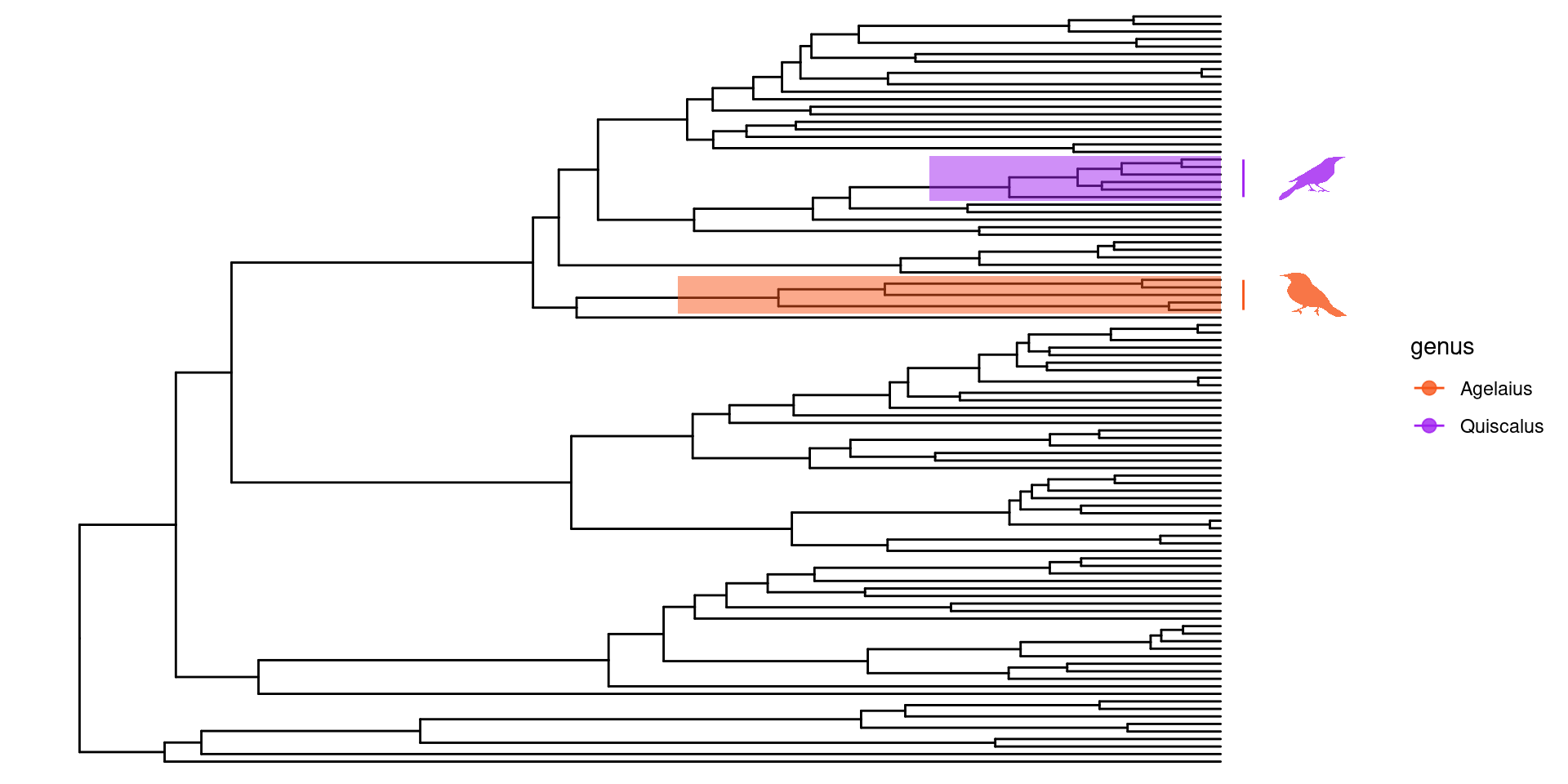
Ahora para la anotación de los árboles, se pueden hacer varias cosas, a mi, por ejemplo, me gusta mucho el género Quiscalus, y quisiera saber donde se encuentra en la filogenia.
Para esto primero debemos encontrar el nodo del ancestro en común más reciente para este grupo. Usando un tibble y la estructura de dplyr es muy facil, primero transformamos nuestro árbol en un tibble y despues utilizando un filtro buscamos las especies de Quiscalus:
Después usando la función MRCA podemos encontrar el ancestro en común de estas especies, en las cuales podemos usar los nombres de las especie con la longitud de rama más alargada y con la longitud de rama más corta, o sus nodos:
Ahora sabiendo que el ancestro en común de Quiscalus se encuentra en el nodo 146, puedo utilizar esta información para anotar la filogenia utilizando el parametro geom_cladelab:
También podemos dibujar una linea entre dos taxa, que pudieran o no estár relacionados, utilizando el parametro geom_strip:
p1+xlim(0,15)+geom_strip("Quiscalus_quiscula","Icterus_icterus",label=" un clado polifilético", barsize =2, offset.text =0.2)
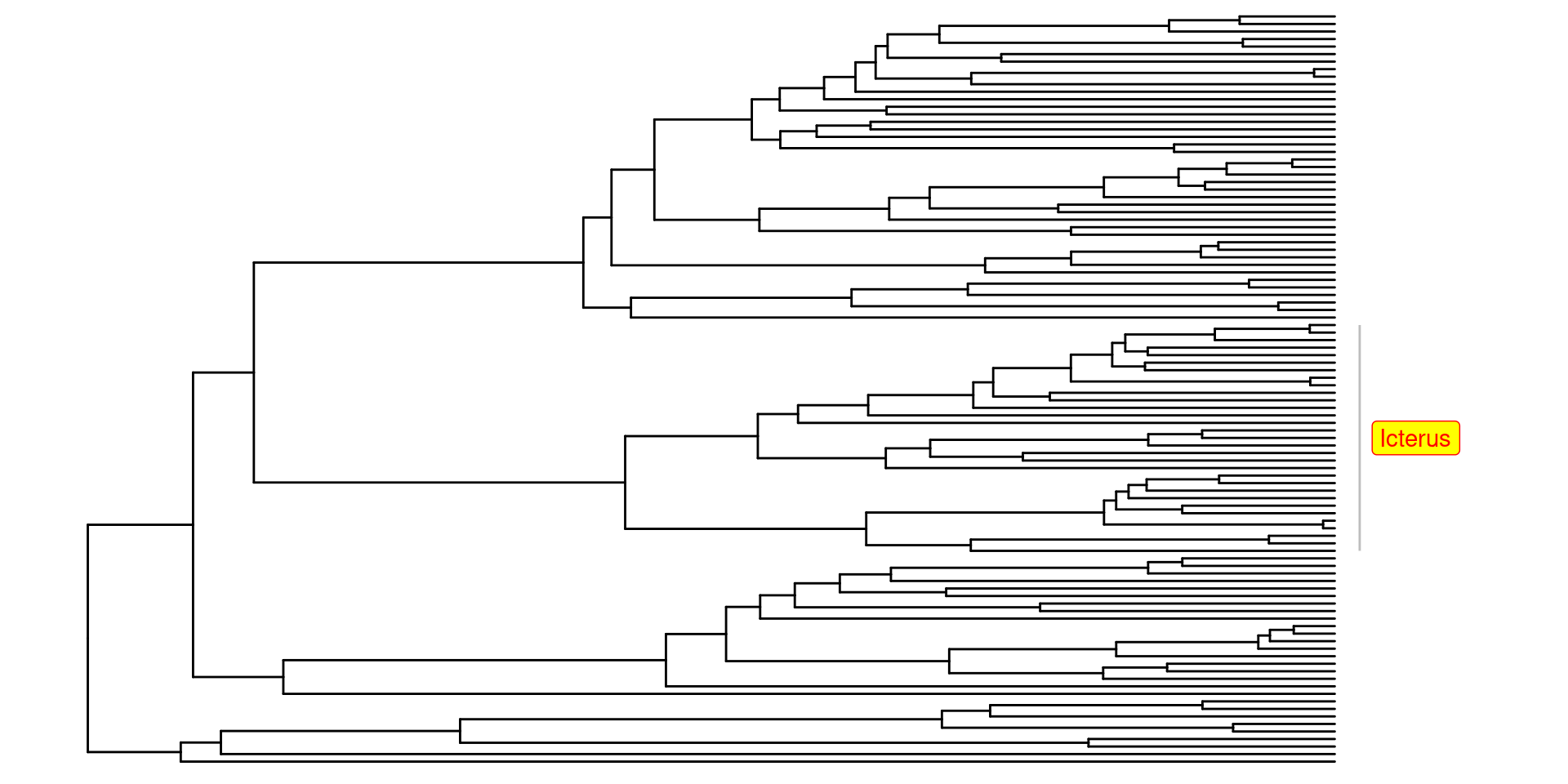
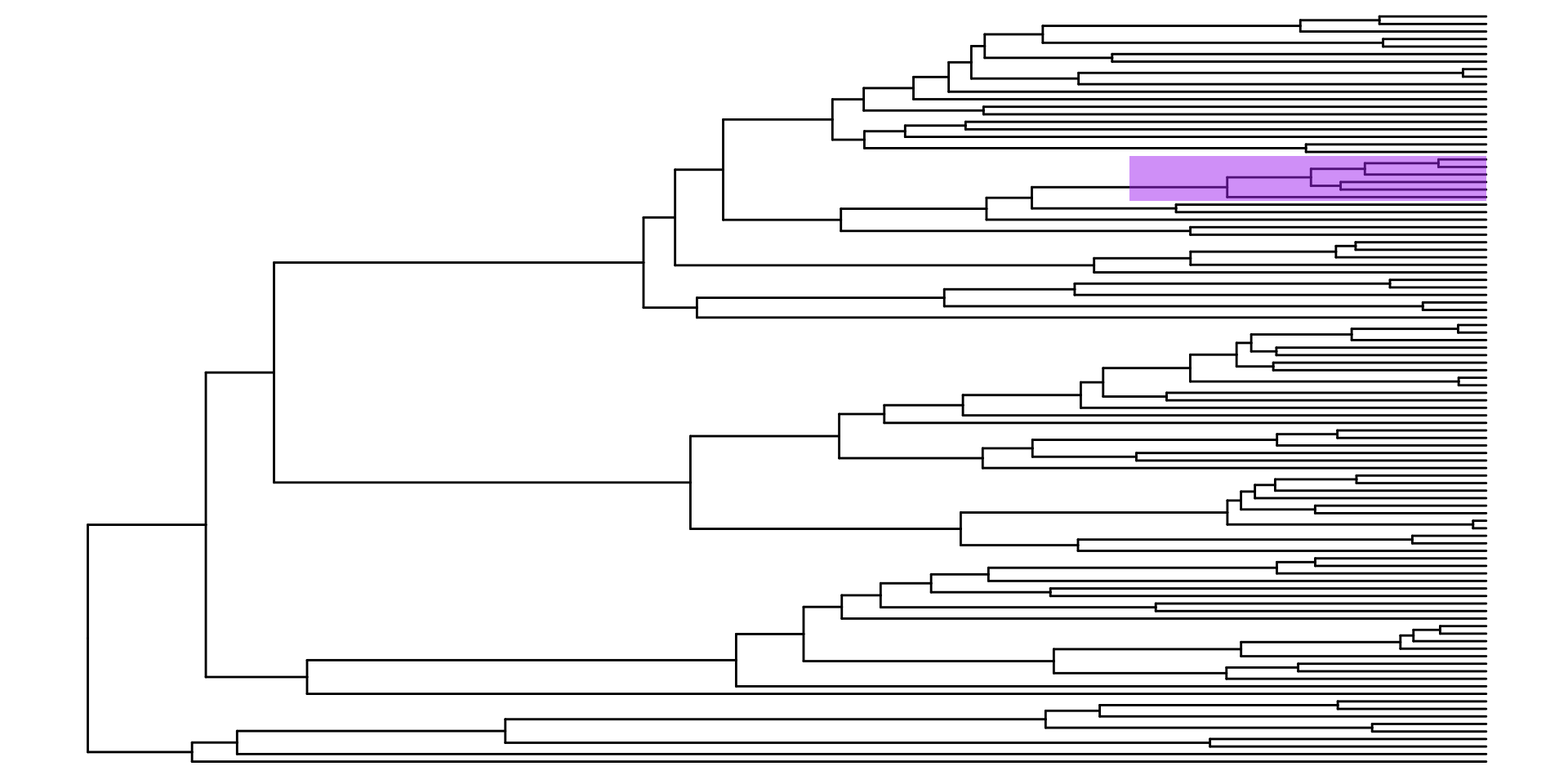
Un parametro de anotación muy bueno, también es el geom_highlight, el cual nos permite destacar clados en particular, utilizando los nodos de ancestro en común:
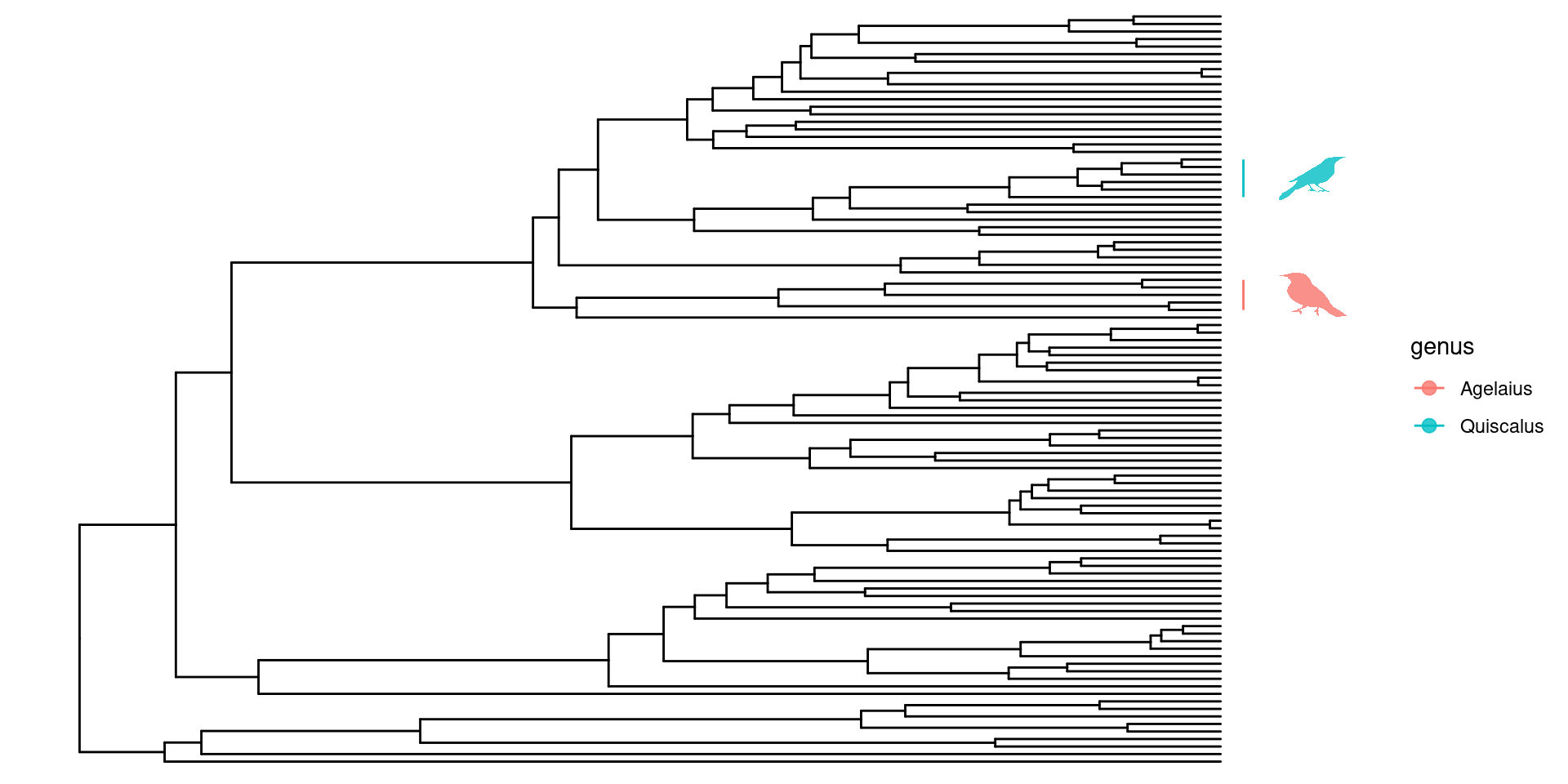
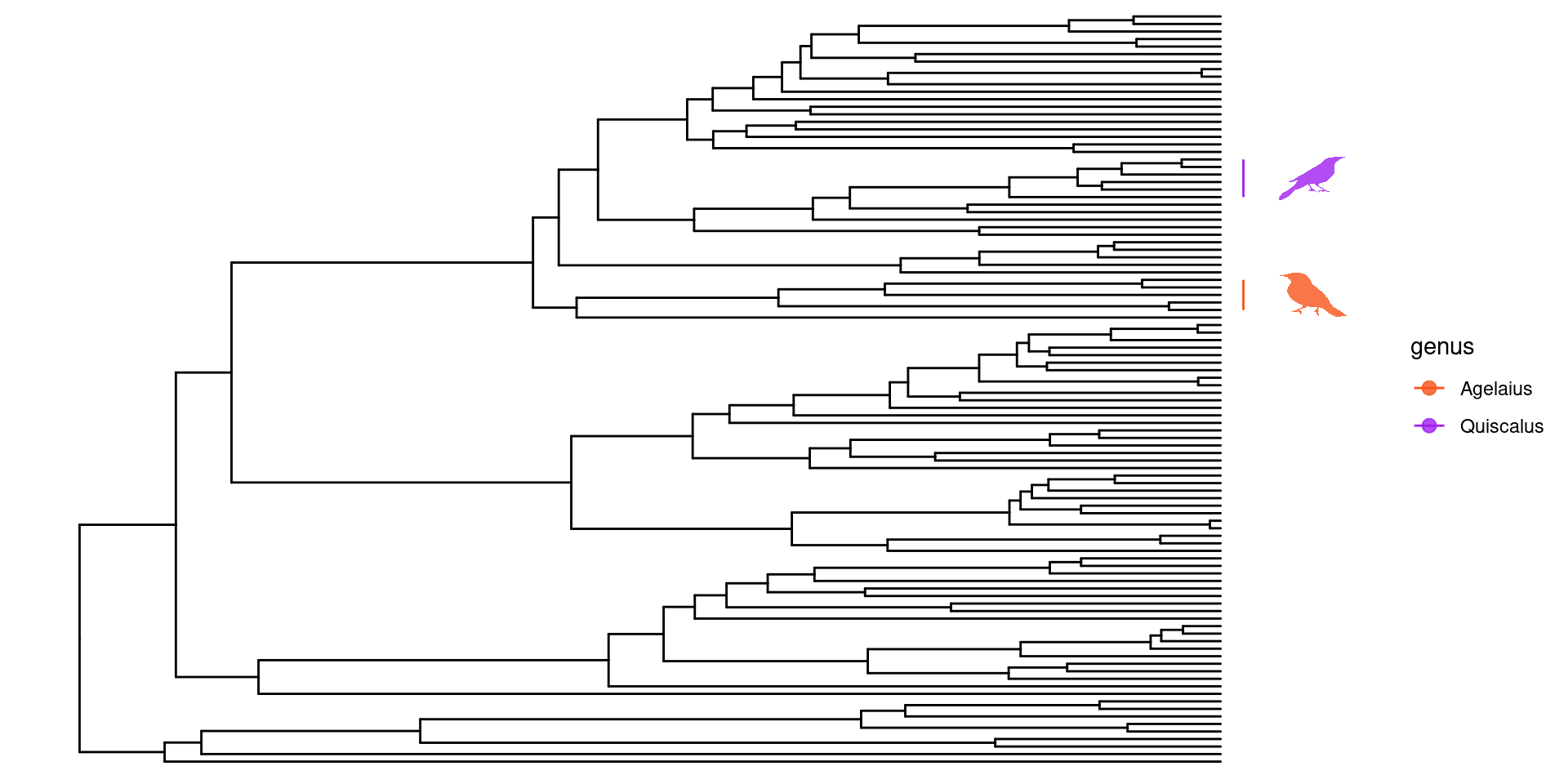
Una función bastante interesante de ggtree es que se pueden personalizar las filogenías utilizando recursos en línea como phylopic o enriquecerlas con imagenes propias. Para poder hacer uso de esta función, primero debemos cargar un paquete extra:
library("rsvg")
Hacer uso de phylopic para personalizar las anotaciones de las filogenias requiere que primero hagamos una tabla con los nodos, el nombre de la especie o clado a los cuales vamos a anotar y el phylopic_id.
En este ejemplo utilizaré los clados Quiscalus y Agelaius, que sé que tienen imagenes indexadas en phylopic. Encontrar los phylopic_id es fácil usando la función phylopic_uid:
ids<-phylopic_uid(c("Quiscalus","Agelaius"))ids
name uid
Quiscalus Quiscalus 2ada4e2f-35ab-48d2-b32e-3bad57033dd4
Agelaius Agelaius 9a3f70a3-2ea9-45aa-8a1a-7c599952fd6c
Con estos ids, ya podemos crear nuestra tabla con los datos necesarios y después gráficar nuestra filogenia:
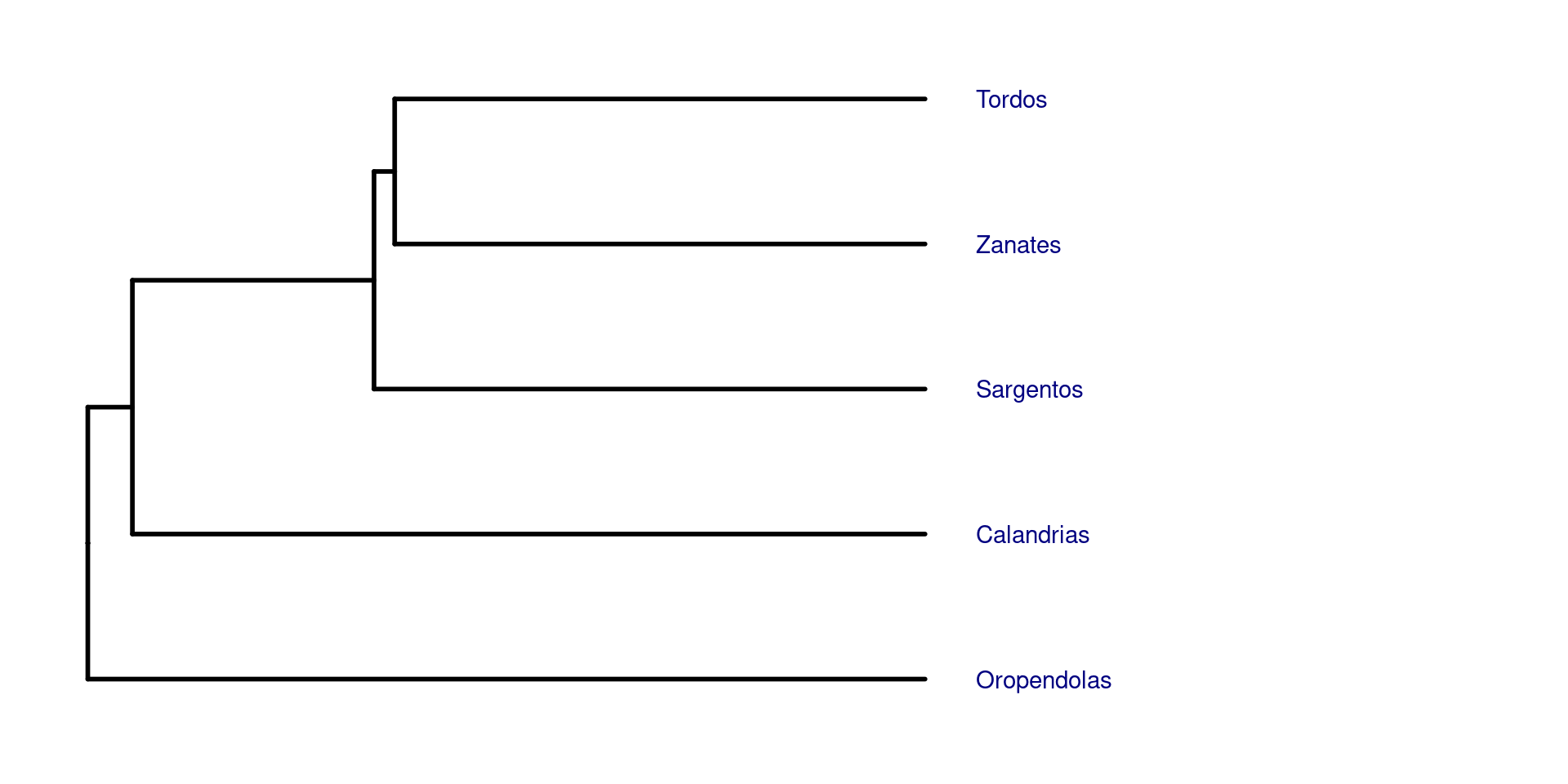
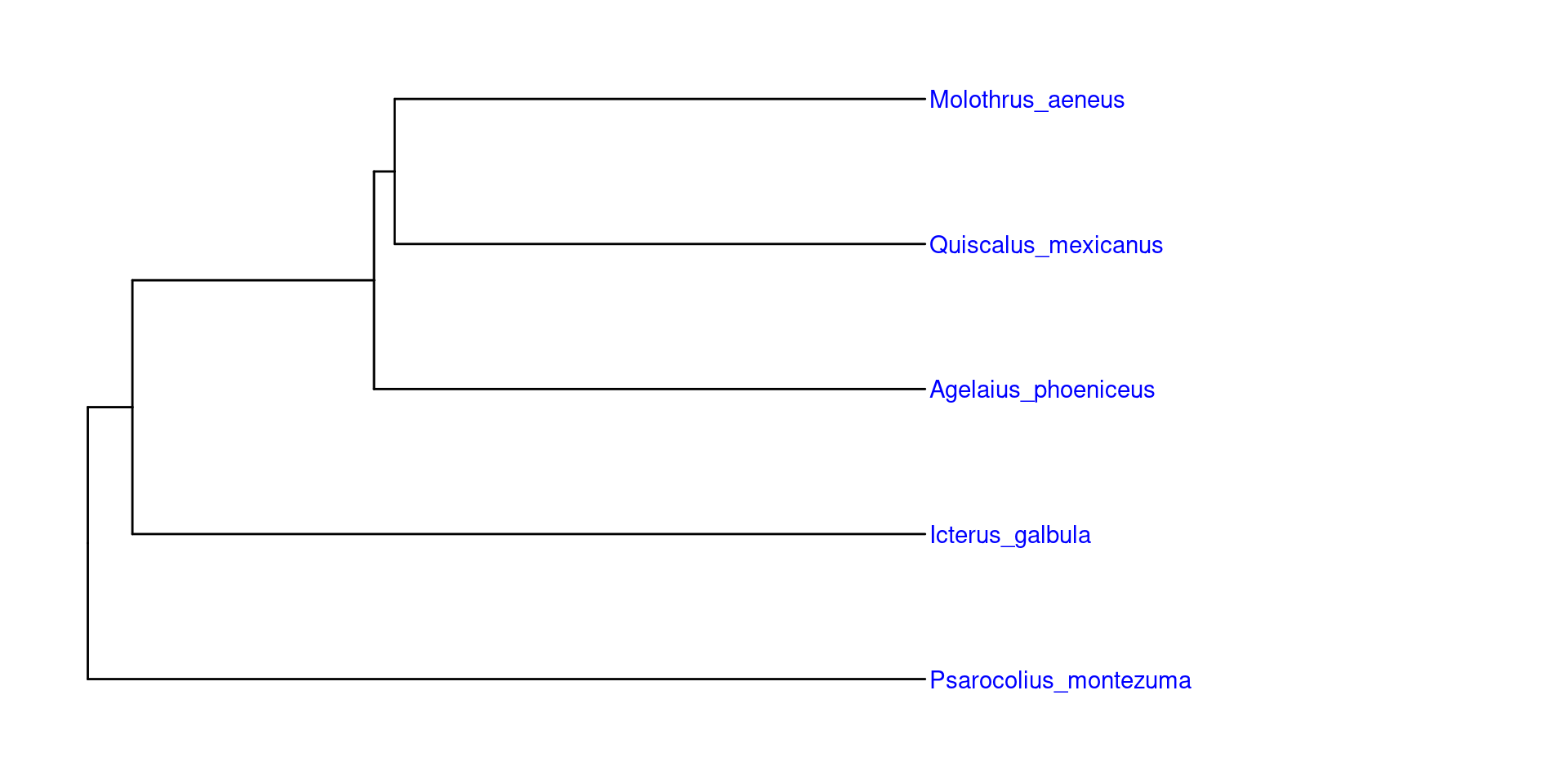
Para poder anotar las filogenias con imagenes, es recomendable usar árboles filogéticos pequeños, en los cuales quizá con un grupo de representantes bastaría. En este ejemplo usaremos una filogenia parafiletica con los generos Quiscalus (Los zanates), Icterus (Las calandrias), Molothrus (Los tordos), Agelaius (Los sargentos) y Psarocolius (Las oropendolas):
Entonces, primero recuperamos un representante de cada grupo, ape es muy bueno para esto usando la función keep.tip:
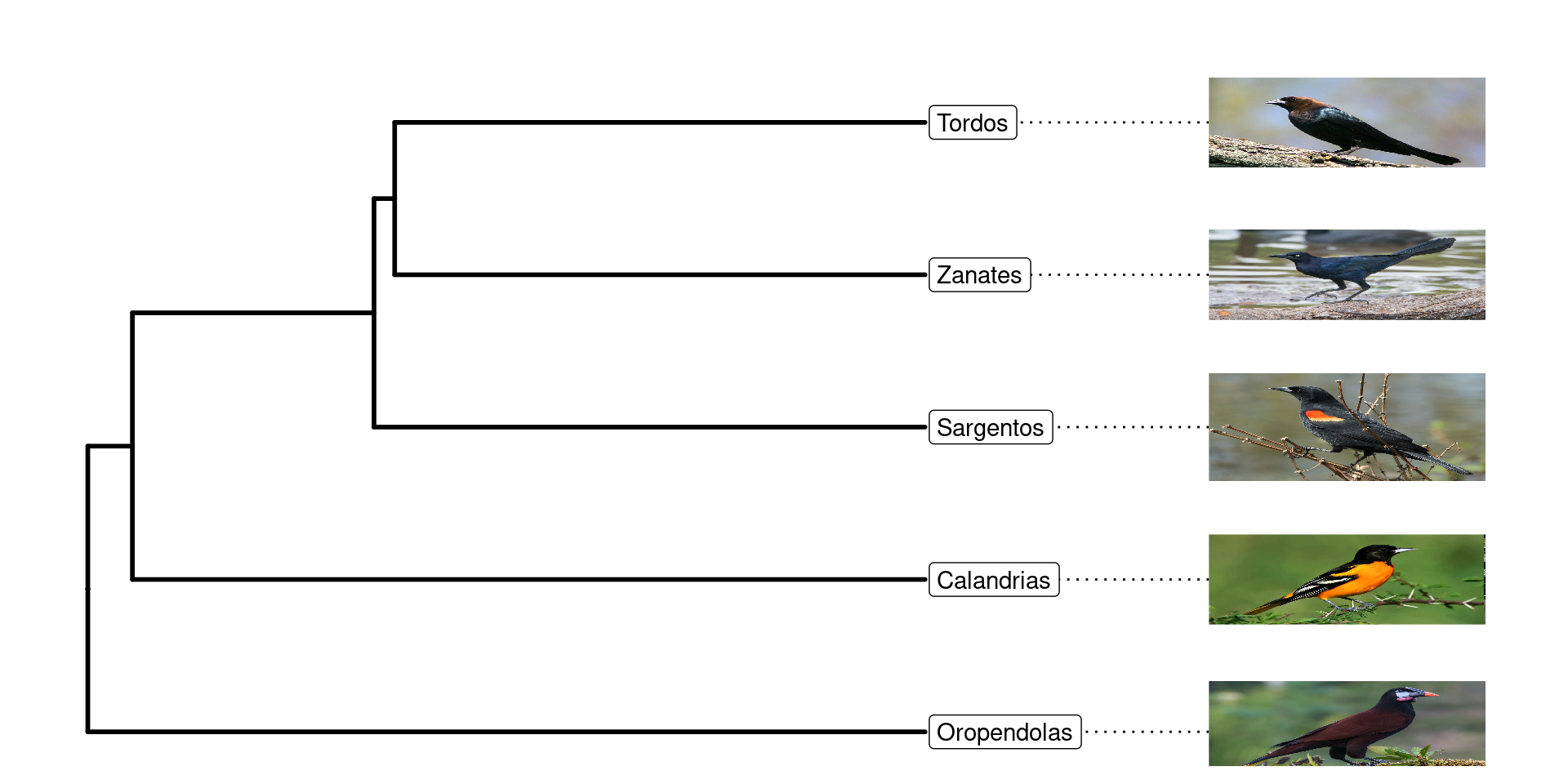
Una vez teniendo este árbol parafilético, podemos colocar las imagenes gusto en sus grupos correspondientes, es importante considerar, que las imagenes deben tener el nombre exacto del grupo o especie y el mismo formato:
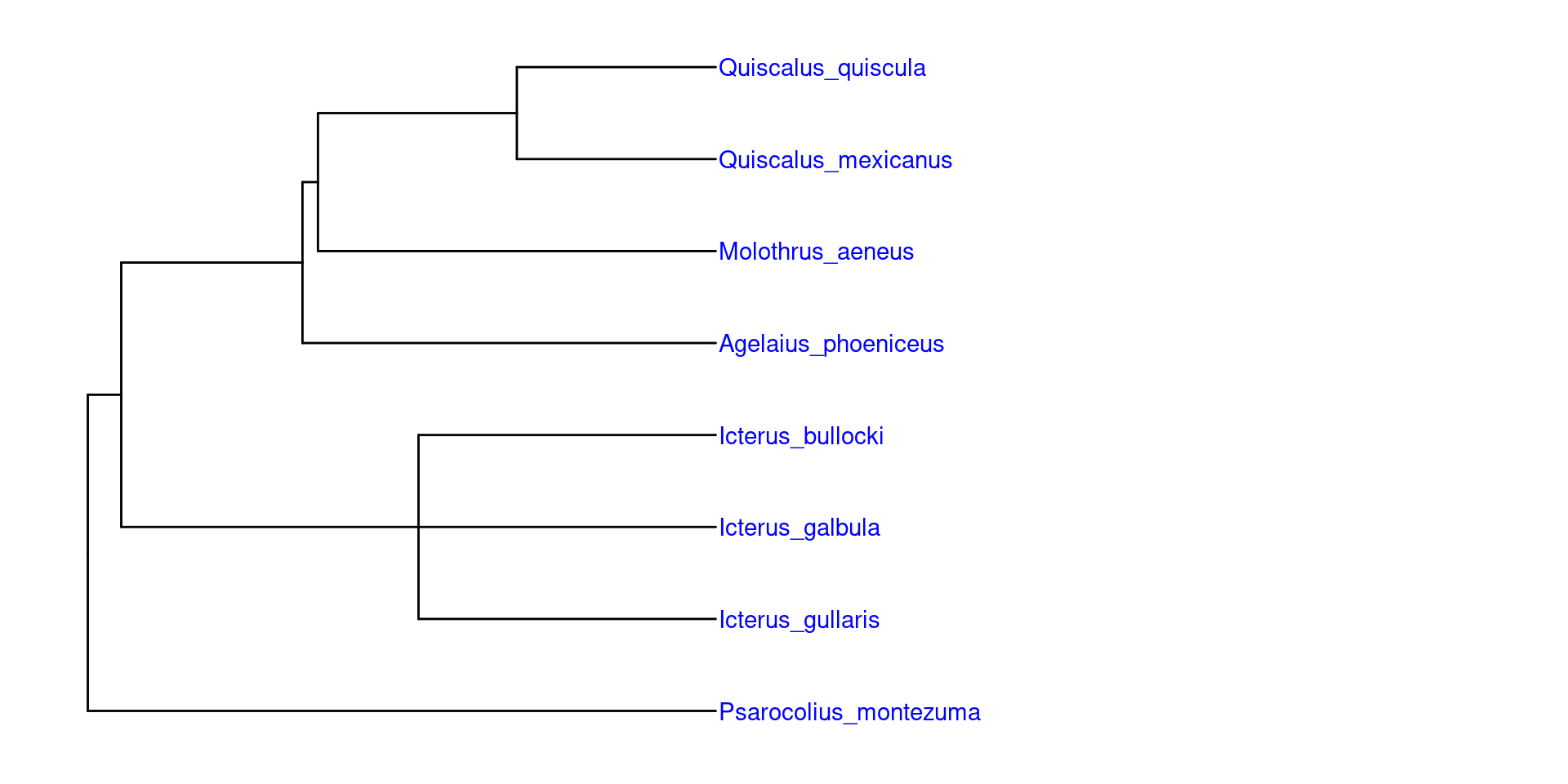
También es posible agregar especies a la filogenia dentro de sus respectivos generos, pero hay que tener cuidado con esto, pues posiblemente nos genere politomías o colocarlas de manera discordante, y esto puede ser problématico para estudios de diversificación o evolución de atributos.
Volveremos a usar nuestra filogenia de grupos y le agregaremos dos especies de Icterus y una especie de Quiscalus
Una vez cargados los datos, la manera más fácil de utilizarlos es uniendolos a la filogenia usando la funcion full_join, es importante que las especies estén etiquetadas como label, para que la función las reconozca:
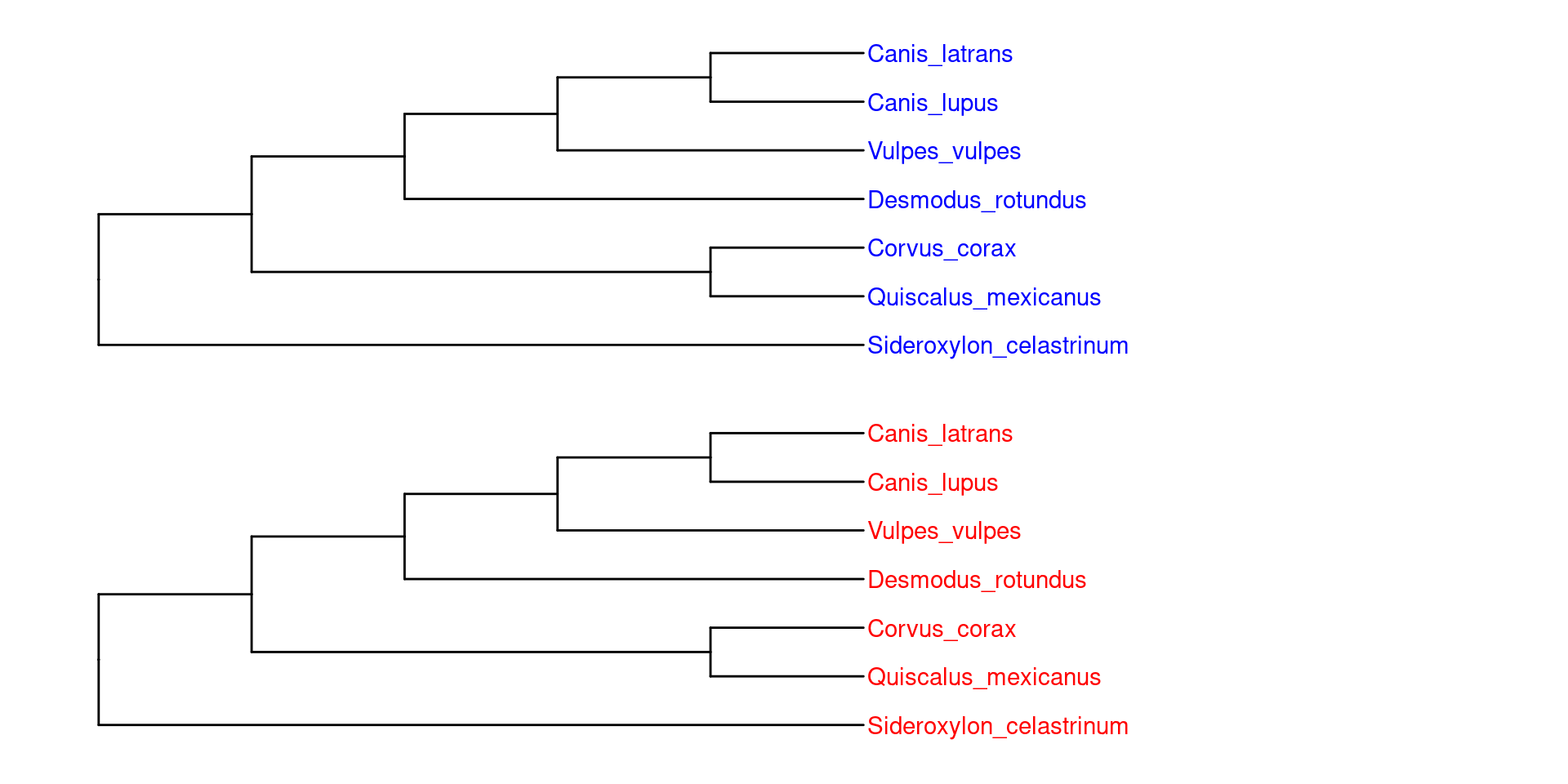
Para la función de rtol, no es necesario introducir las especies en orden, ya que ésta recuperará la taxonomia válida y nos arrojará una filogenia podada.
En este caso, OTL nos regresó exactamente la misma filogenia que creamos al principio: